-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Advances in Analytical Chemistry
p-ISSN: 2163-2839 e-ISSN: 2163-2847
2013; 3(2): 15-19
doi:10.5923/j.aac.20130302.02
Method Development and Validation of Carisoprodol and its Impurities by Ultra Violet-High Performance Liquid Chromatography
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLT. Rohith1, S. Ananda1, Netkal M. Made Gowda2
1Department of Studies in Chemistry, Manasagangothri, University of Mysore, 570009, Mysore
2Department of Chemistry, Western Illinois University, One University Circle, Macomb, 61455, USA
Correspondence to: S. Ananda, Department of Studies in Chemistry, Manasagangothri, University of Mysore, 570009, Mysore.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Ultraviolet-high performance liquid chromatographic (UV-HPLC) method was developed for the quantitation of carisoprodol and its impurities viz 2-methyl-2-propylpropane-1, 3-diyl dicarbamate (impurity-D) and N-isopropyl -2-methyl-2-propyl-3-hydroxy propyl carbamate (impurity-B) in active pharmaceutical ingredient. Validation of the method showed excellent sensitivity, selectivity, accuracy, precision and ruggedness. Efficient chromatographic separation was achieved on a Zorbax eclipse XDB C (8) 250 mm X 4.6 mm id, 5µm , stationary phase in gradient mode and quantitation by ultraviolet wavelength detection. The method was validated as per International Conference of Harmonization (ICH) guidelines in terms of Quantitation limit (QL), Detection limit (DL), Linearity, Precision, Accuracy and Specificity. The QL and DL values for impurity-B were found to be 0.015% and 0.008% and for that of impurity-D were found to be 0.26% and 0.13%respectively, with respect to sample concentration. The method was linear within the range of QL to 200% for the two impurities. Thus, the newly developed method was found to be accurate, efficient and stable. The characterization of these impurities was carried out for the confirmation of respective structures using nuclear magnetic resonance spectroscopy (NMR).
Keywords: Validation, Carisoprodol, UV-HPLC, Impurities
Cite this paper: T. Rohith, S. Ananda, Netkal M. Made Gowda, Method Development and Validation of Carisoprodol and its Impurities by Ultra Violet-High Performance Liquid Chromatography, Advances in Analytical Chemistry, Vol. 3 No. 2, 2013, pp. 15-19. doi: 10.5923/j.aac.20130302.02.
Article Outline
1. Introduction
- Carisoprodol is a centrally acting muscle relaxant, with analgesic properties, making it a popular drug of abuse. Carisoprodol is a dicarbomate, centrally acting, oral skeletal muscle relaxant whose chief application is in the treatment of acute muscular spasm associated with craniomandibular disorder, lumbago, sciatica, and other lower back syndromes[1]. Carisoprodol chemical name being N-isopropyl-2-methyl-2-propyl-1, 3-propanedioldicarbamate, is a pharmaceutical active agent whose metabolite is meprobamate[2]. Carisoprodol, a synthetic compound first synthesized in 1959 is related structurally to meprobamate. Carisoprodol is marketed as a muscle relaxant and dispensed by prescription under the trade names of Soma®. Carisoprodol is indicated for the relief of pain associated with acute musculoskeletal condition and in the treatment of acute muscular spasm[3, 4]. Carisoprodol produce weak anticlolinergic, antipyretic and analgesic effects[4]. The effect of carisoprodol, which includes sedation, loss of balance, confusion, and increased reaction time are similar to those of alcohol, benzodiazepines and other CNS depressants and well documented to decrease human performance and adversely affect driving effect[5]. The structure of carisoprodol is such that it does not have UV chromophore with significant absorbance. Therefore, the USP Assay method for Carisoprodol tablets employs a liquid chromatography equipped with a refractive index detector [6].Our laboratory does not currently have an operational refractive index detector. Official monographs available for carisoprodol drug substance refer to TLC method for impurity estimation[6]. Several methods were reported based on titrimetry, infrared, nuclear magnetic resonance (NMR), gas chromatography, liquid chromatography (LC) and gas chromatography-mass spectrometry (GC-MS) for the determination of carisoprodol analysis[8-20].This paper describes the UV-LC method for determination of carisoprodol and its impurities. Developed method was validated as per ICH guidelines. Chemical structure of Carisoprodol and its impurities are shown in figure 1- figure 3.
1.1. Chemical Structures of Carisoprodol and its Impurities
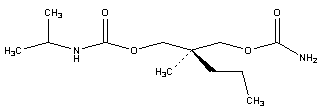 | Figure 1. (2RS)-2-[(Carbamoyloxy) methyl]-2-methylpentyl (1-methylethyl) carbamate (Carisoprodol) |
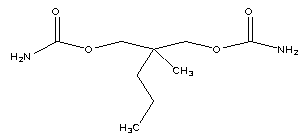 | Figure 2. 2-methyl-2-propylpropane-1, 3-diyl dicarbamate (impurity-D) |
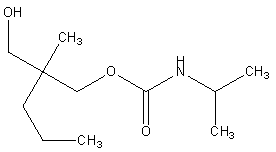 | Figure 3. N-isopropyl-2-methyl-2-propyl-3-hydroxy propyl carbamate (impurity-B) |
2. Experimental
2.1. Reagents and Samples
- HPLC grade Acetonitrile was purchased from Merck, water used was from a milli-Q purified system, Millipore, Orthophosphoric acid from Merck and triethyl amine was purchased from spectrochem. Carisoprodol of sample purity 99.18% was used in this study. Process related impurities, that is-Impurity-B and Impurity-D were obtained from Chemical Research and Development Department, Troy Life Sciences, Bangalore.
2.2. High Performance Liquid Chromatographic Conditions
- The HPLC system consisted of a Shimadzu prominence separate module LC-10AD equipped with PDA detector water Empower-2 software was used for the data acquition and processing. Zorbax eclipse-XDB C (8) HPLC column of 250 mm X 4.6 mm id, 5µm particle size was used. The column was maintained at 30℃±2℃. The buffer preparation was by dissolving (0.1M orthophosphoric acid) 5.5ml of orthophosphoric acid in 950ml of water, pH adjusted to 3.1 by using triethylamine. Mobile phase pump-A was mixture of buffer and acetonitrile in the ratio (67:33v/v) and mobile phase pump-B was mixture of buffer and acetonitrile in the ratio (33:67v/v). The flow rate was set at 1.0ml/min and UV detector 200nm.The injection volume was 20µl.The gradient elution was (Tmin A: B) T0100:0, T25100:0, T3070:30, T4050:50, T45100:0, T52100:0.The diluent used was mobile phase-A throughout the analysis.
2.3. Preparation of Stock Solution for Method Validation
- A test preparation of 9 mg/ml of Carisoprodol API sample was prepared by dissolving in diluent (mobile phase-A). A stock solution of impurity-D was prepared by dissolving 90mg standard sample in to100 ml volumetric flask and made up to the 100ml with diluent. 5ml of above solution was transferred into a 100ml volumetric flask and made up to the volume with the diluent. The standard solution of impurity-D was prepared at 0.5% with respect to sample concentration (9mg/ml). A stock solution of impurity-B was prepared by dissolving 90mg standard sample in to 10 ml volumetric flask and made up to the 100ml with diluent. 2ml of above solution was transferred into a 100ml volumetric flask and made up to the volume with the diluent. The standard solution of impurity-B was prepared at 0.2% with respect to sample concentration (9mg/ml).
3. Results and Discussion
3.1. Method Development
- Carisoprodol lacks any appreciable UV absorbance or fluorescence. Therefore, GC-MS methods are mainly used. However, problems encountered with GC are due to the heat instability of carisoprodol at the injection port, leading to thermal decomposition which results in poor chromatography. Hence, it was necessary to develop a rugged method of UV-HPLC in order to overcome such problems. Various methods, stationary phases, diluents were used for the development of sensitive and accurate method for carisoprodol and its impurities. HPLC with UV detection was chosen as simple, fast and effective separation method for determination of Carisoprodol and its process related impurities. All compounds were tested at different wavelength and had a low detector response for detection of the compounds, however, 200nm was chosen especially with regard to absorption spectra of Carisoprodol and its impurities, both gave higher detector response at 200 nm, therefore the final absorption wavelength for detection was chosen at 200 nm. Several analytical columns were chosen and tested during the development of this method. GL-Science inertsil ODS column, waters X-Terra column, Agilent Zorbax reversed-phase bonded phases based on ultra-pure silica-Stable bond (SB)-provided poor peak shape and sample resolution, as well as no long column lifetimes. Different mobile phases consisting of acetate buffer, formate buffer, mixture of Acetonitrile and water were tested with different pH. Numerous trials were carried out with different sample preparation solutions to enhance peak responses. For instance methanol, acetonitrile, mixture of acetonitrile and water. Even derivatizations of samples were carried out using picrate derivative and benzyl derivative. Sample solutions were also prepared by mixing methanol: perchloric acid: triethyl amine and acetonitrile: perchloric acid: triethyl amine. Also with above mentioned combinations, the solution was heated at 30℃ for few minutes. Even after so many experiments it was hard to optimize the HPLC method. Finally, best results were obtained using a buffer consisting of 5.5ml phosphoric acid in 950 ml water, pH adjusted to 3.1 by using triethyl amine along with Zorbax eclipse XDB C (8) column (250 X 4.6mm 5um) and hence were found suitable for the analysis. Mobile phase-A consisted mixture of buffer and acetonitrile in the ratio (67:33 v/v) and mobile phase-B consisted mixture of buffer and acetonitrile in the ratio (33:67 v/v). Ultimately, sample was prepared by using mobile phase-A. Thus, a significant HPLC method was developed and optimized. In this method Impurity-B RRT was about 2.29, and impurity-D RRT was about 0.31.Figure 4 and 5 depicts the experimental chromatograms of spiked solution of carisoprodol with impurities and QL solution respectively.
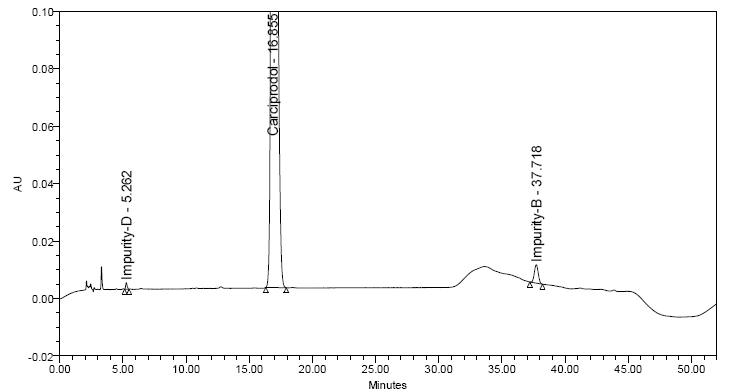 | Figure 4. Spiked solution of Carisoprodol and its impurities |
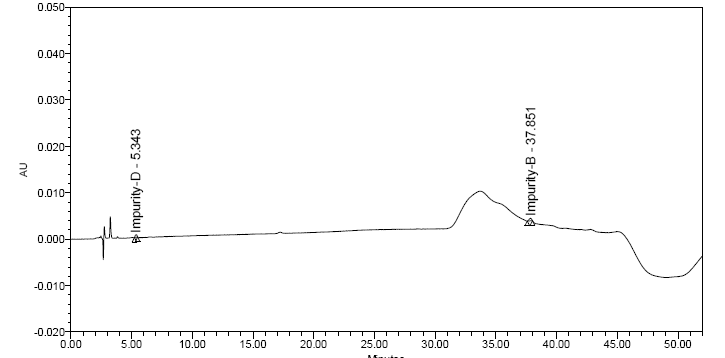 | Figure 5. LOQ Chromatogram of Carisoprodol impurities |
|
4. Method Validation
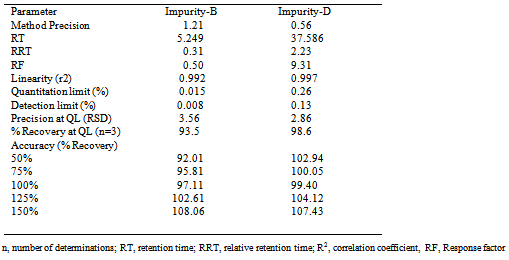
- A newly developed and optimized method was validated for Quantitation limit (QL), Detection limit (DL), Linearity, Precision, Accuracy, Specificity and Robustness as per ICH guidelines. Validation was carried out for two process related impurities, viz. impurity-B and impurity-D. Validation study was carried out for impurity-B and impurity-D. The selectivity was checked by injecting 9mg/ml of Carisoprodol solution containing 0.2% of impurity-B and 0.5% of impurity-D monitored throughout the validation. Method validation results are summarized in table 1.
4.1. Specificity
- To demonstrate the specificity of HPLC method, in all the impurity spiked samples, the purity angle obtained for Carisoprodol and impurity peaks was less than purity threshold demonstrating spectral homogeneity. During this study impurity-B and impurity-D got well separated from each other and as well as from Carisoprodol which proved that the adopted method was specific.
4.2. Linearity, RRF, Detection limit (DL) and Quantitation limit (QL)
- The linearity was established by measuring area responses for impurity-B and impurity-D, linearity ranging from QL to 200% with respect to sample concentration (9mg/ml).Seven concentrations were prepared across the range and injected in triplicates. The average area calculated was plotted against the concentration. The correlation co-efficient obtained was greater than 0.99 for impurity-B and impurity-D. The results are presented in table-1. The Quantitation limit (QL) and Detection limit (DL) for Carisoprodol impurities were determined by signal to noise ratio method.
4.3. Precision and Accuracy
- The precision of the related substance method was checked by injecting six individual preparations of (9 mg/ml) Carisoprodol spiked with 0.2% of impurity-B and 0.5% of impurity-D. Percentage RSD for peak areas of each impurity was calculated and study was also performed in the same procedure on a different day. The intermediate precision of the method was also evaluated by a different analyst and different instrument in the same laboratory. Percentage RSD of areas of each impurity was less than 5.0, confirming good precision. Accuracy was validated through recovery experiments by spiking known amount of impurity (50%, 75%, 100%, 125% and 150%) with Carisoprodol with respect to sample concentration (9mg/ml). Each parameter was analyzed in triplicates and percent recoveries were calculated. The results of accuracy and precision are shown in table-1.
4.4. Solution Stability and Mobile Phase Stability
- Required analysis was carried out regarding solution stability and mobile phase stability. It was observed that solution of impurity-D and Carisoprodol was found to be stable up to 72 hrs unlike impurity-B solution which was stable only up to 24 hrs.
5. Conclusions
- Hence method suggested was found to be simple, accurate, selective and equally sensitive. The method was fully validated showing satisfactory data for all the method validation parameters tested. The developed method can be conveniently used by quality control department to determine the related substances in regular Carisoprodol production samples.
ACKNOWLEDGEMENTS
- The authors wish to thank the management of Troy Life Sciences Private Limited, Dr. Rakesh sahay and Anvita G.P, for their constant support and encouragement providing necessary facilities to carry out this work.