-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Advances in Analytical Chemistry
p-ISSN: 2163-2839 e-ISSN: 2163-2847
2012; 2(4): 25-31
doi: 10.5923/j.aac.20120204.01
Enantioseparation of Nadifloxacin by High performance liquid Chromatography
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLSurendra Dutt Sharma , Gaurav Singh
Analytical Research Laboratory, School of Sciences.IFTM University Campus, Moradabad 244102, Uttar Pradesh, India
Correspondence to: Gaurav Singh , Analytical Research Laboratory, School of Sciences.IFTM University Campus, Moradabad 244102, Uttar Pradesh, India.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
A rapid isocratic chiral HPLC method has been developed for the separation of R-Nadifloxacin from S-Nadifloxacin. Good resolution viz. Rs> 4.0 between R- and S- forms of Nadifloxacin was achieved by RP-HPLC using endcappedC18 stationary phase and chiral mobile phase. Chirality to the mobile phase was imparted with addition of β-CD in phosphate buffer with EDTA.Column temperature was 45°C and flow rate was kept 1.5 mL min-1.The elution was monitored by UV-vis detector at λ-290 nm. The calibration curve showed excellent linearity over concentration range 0.040-20 µg mL-1.This method was further used to determine the amount of R-Nadifloxacin in pure and active pharmaceutical ingredient of S-Nadifloxacin and is capable to quantitate and detect R-Nadifloxacin to the levels of 0.040µg mL-1and 0.020µg mL-1 respectively. The average recovery of R-Nadifloxacin was 99.09 %. The method is better than the already reported one for the enantioseparation of the Nadifloxacin.
Keywords: Enantioseparation, Chiral Mobile Phase Additive, (R,S)-Nadifloxacin, β-Cyclodextrin
Article Outline
1. Introduction
- In recent years, there has been considerable interest in the synthesis and separation of enantiomers of organic compounds especially because of their importance in biochemistry and pharmaceutical industry[1-5]. The method frequently used for separations, monitoring the progress of an asymmetric synthesis or optical purity of the product is chromatography with either liquids, gases or supercritical fluids as the mobile phase. Separation of the enantiomers comprising the racemate i.e. the resolution of the racemate is a common problem in stereochemical research as well as in the preparation of biologically active compounds, in particular drugs. One approach to separate enantiomers sometimes referred to as indirect enantiomeric resolution which involves the coupling of the enantiomers with an auxiliary chiral reagent to convert them in to diastereomers[6-11]. The diastereomers can be separated by any achiral separation technique[1]. The chirality of the enantiomeric molecules is caused by the presence of one or more chiral atom in their structure. The chirality sense and optical activity of the enantiomers are determined by their absolute configuration. Reversed phase high performance liquid chromatography (RP-HPLC) techniques were used to separate and quantify enantiomers with high resolution.(S)-(-)-Nadifiloxacin[(s)-(-)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidino)-5-Methyl-1-oxo-1H,5H-benzo[i,j]quinolizine-2-carboxylicacid] is a synthetic quinolone derivative with potent broad spectrum antibacterial activity and inhibits the enzyme DNA gyrase that is involved in the synthesis of bacterial DNA and its replication[12].
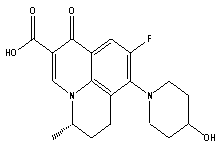 | Figure 1a. Chemical structure of S-Nadifloxacin |
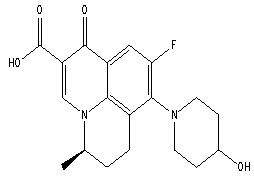 | Figure 1b. Chemical structure of R-Nadifloxacin |
2. Experimental
2.1. Instrumentation
- Chromatographic analysis is performed with waters 2695-separation module coupled with photodiode array detector (Compounds were detected at λ-290 nm) and column YMC C18 (250 mm x 3.0 mm) 3 μm pore size 120 Ă is used for chromatography. The oven temperature of HPLC was at 45°C and injection volume is 20 μL. The flow rate of the mobile phase was adjusted at 1.5 mL min-1 and the total run time is 30 min. Chromatographic data were controlled and processed on a computer running with millennium empower pro version 5.00.00.00.
2.2. Chemicals
- Nadifloxacin is from Beijing Nine camp Medicine Technology Co.Ltd. Chaoyang (CHINA), Sodium hydroxide pellets, Triethylamine, di-sodium hydrogen orthophosphate (anhydrous) and Potassium di-hydrogen orthophosphate (all of AR grade); water and acetonitrile (HPLC grade) are from qualigens (India). Ethylene diaminetetraacetic acid (EDTA) disodium salt and β-Cyclodextrin (both of AR grade) from Across chemicals (India) were used during studies.
2.3. Preparation of Stock Solution for Resolution
- 5.0 mg of R-Nadifloxacin is weighed accurately and transferred into a standard 50 mL volumetric flask. It is thoroughly dissolved with 2 mL of 0.1 N NaOHsolution and the volume was made up with diluent up to the mark and mixed thoroughly.
2.4. Preparation of Solution for Resolution
- 50 mg of S-Nadifloxacin is weighed accurately and transferred in to 25 mL volumetric flask. It is dissolved with 2 mL of 0.1 N NaOH and 0.5 mL of resolution stock solution is added. The solution is now made up with diluent up to the mark and thoroughly mixed. It is then filtered through 0.45µm filter or finer porosity membrane filter.
2.5. Preparation of Sample Solution
- 50 mg of S-Nadifloxacin is weighed accurately and transferred in to 25 mL volumetric flask. It is dissolved with 2 mL of 0.1 N NaOH. The solution is now made up with diluent up to the mark and thoroughly mixed. It is then filtered through 0.45µm filter or finer porosity membrane filter.
2.6. Preparation of Buffer
- 1.0 g of Potassium di-hydrogen orthophosphate, 0.50 g of di-Sodium hydrogen orthophosphate (anhydrous), 50.0 mg of E.D.T.A and 10.0 g cyclodextrin hydrate is dissolved in 1000 mL of water by constant stiring. It is then filtered through a 0.45µm filter or finer porosity membrane filter.
2.7. Preparation of Mobile Phase
- A mixed degassed solution of buffer and acetonitrile (ACN) in various ratios (v/v) is prepared and its pH is adjusted to the desired level with triethylamine. The mobile phase is also used as diluent.
3. Results and Discussion
- Enantiomeric separation by HPLC is generally made by immobilizing the single enantiomers on to the stationary phase and resolution is the result of the formation of transient diastereomer by the interaction of CSPs. Enantiomer which form the most stable diastereomer is retained and opposite enantiomer forming less stable diastereomer will elute first. Interactions between CSPs and enantiomers are very week and require careful optimization by adjustment of the suitable mobile phase and temperature of the column to maximize the enantioselectivity. These interaction forces are ionic, π-π interaction, hydrophobic effect and hydrogen bonding[6].For cyclodextrin produced by the action of Bacillus macerans amylase on starch, the size of CD depends upon the type of reaction between the two. CDs are the cyclic oligosaccharides containing six to twelve or eight D (+) glucopyranose units and bonded through alpha 1-4 linkages and form truncated conical cavity, the diameter of which depends upon the number of glucopyranose units. Commercially available CDs are pictured as hollow truncated cones, the torrodial structure having a hydrophilic surface. β-phase has seven units of glycopyranose the value ranging from (6.0-8.0 Ă). β-CD has the widest application due to its pronounced kink shape whereas α-CD and γ-CD are more planer. Its molecule has secondary -2 and -3 hydroxyl groups of the glycopyranose units lining at the mouth of the cavity and primary 6-hydroxyl groups at the rear of the cavity (fig-2).
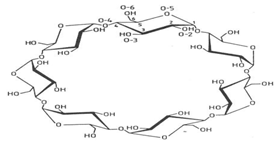 | Figure 2. Structure of β-Cyclodextrin |
4. Optimization of the HPLC Enantioseparation
- The main features of the enantioseparation of racemic mixture of Nadifloxacin are given below. Tables 1-3 shows the effect on the USP (United States Pharmacopeia) resolution of the variation in the composition of the mixture of buffer and acetonitrile alongwith variation in final pH value and the flow rate of the mobile phase.
|
|
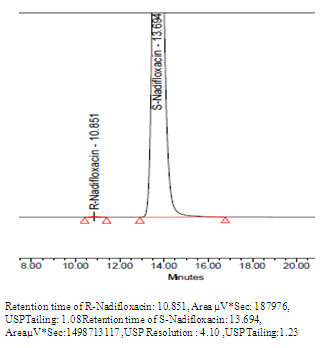 | Figure 3. Chromatogram of S-Nadifloxacin spiked with R-Nadifloxacin |
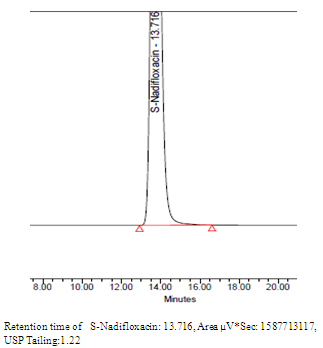 | Figure4. Chromatogram of S-Nadifloxacin |
5. Validation of Analytical Method
- After the systematic optimization of the method the final conditions were found to be as follows: mobile phase (buffer and ACN) ratio is 90:10, pH 7.4, flow rate 1.5 mL min-1 and total run time is 30 min.
5.1. System Suitability
- Performance of the method was determined by injecting the resolution solution. The qualification criteria were resolution between two enantiomers which should be not less than 2.0 and tailing factor not more than 1.5, which ensures baseline separation and symmetrical peak shape for R-Nadifloxacin.
5.2. System Precision
- The System precision of the analytical method was determined by injecting the replicate injection of R-Nadifloxacin with concentration 2 µg mL-1.
5.3. Method Precision
- Precision of the method was determined by the analysis of six spiked sample amount of R-Nadifloxacin at concentration of 2 µg mL-1 shown in (table -4).
5.4. Ruggedness
- Ruggedness of the method was determined by the analysis of six spiked sample amount of R-Nadifloxacin at concentration of 2 µg mL-1 on different instrument and different column shown in (table -4).
5.5. Accuracy
- Accuracy of the method was confirmed by determining the % recovery of spiked amount ofR-Nadifloxacin at concentration 80, 100 and 120 % of 2 µg mL-1 in thepre-analyzed sample of Nadifloxacin and is shown in (table -5).
| |||||||||||||||||||||||||||||||||||||||||||||
|
5.6. Linearity of R-Nadifloxacin
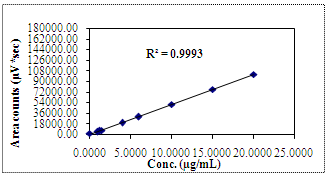 | Figure 5. Linearity Curve of R-Nadifloxacin |
5.7. Limit of Detection and Quantification
- The limits of detection and quantification of R-Nadifloxacin in this method are summarized (table-6). As per Eurochem guidelines, the % RSD of six replicate injection peak area should be not more than 10 for LOQ. In case of LOD, the % RSD of six replicate injection peak area should be not more than 33. The limit of quantification and limit of detection for R-Nadifloxacin are 0.040 µg mL-1 and0.020 µg mL-1 respectively.
|
6. Conclusions
- A simple, rapid and linear RP-HPLC method containing chiral mobile phase additive is given for the quantitative separation of R-Nadifloxacin from S-Nadifloxacin. The method is simple as it does not need any derivatization to diastereomers and economical too. It excludes the use of chiral stationary phase and uses very inexpensive β-cyclodextrin as chiral mobile phase additive and easily available RP-HPLC C18 column for chromatography.
ACKNOWLEDGEMENTS
- The authors thank Ankitasingh for editing the manuscript and organizing it for Web submission.
References
| [1] | R.D. Yeole, A.S. Jadhav, K.R. Patil,V.P. Rane, M.L. Kubal, S. Singh ,M.V. Patel and H.F. Khorakiwala, “Validated chiral high-performance liquid chromatography method for a novel anti-methicillin-resistant staphylococcusaureusfluoroquinolone WCK 771”, J. chromatogr. (A), Vol.1108, pp.38-42, 2006. |
| [2] | T.Ooya, N.Kobayashi, T.Ichi, S. Sasaki and N.Yui, “Hydrogels having tubular α-cyclodextrin structure: effect of nano-tube structure on long alkyl chain partitions”, Science and Technology of Advanced Materials, Vol.4, pp. 39-42, 2003. |
| [3] | E.Lipika, A.Selouane, D.Postel, C.Len, M.P.Vaccher, J-P.Bonte and C.Vaccher, “Enantioseparation of cis trans diastereomers of 2’3’-didehdro-2’,3’dideoxythiamidine analogs by high performance liquid chromatography and capilary electrophoresis”, J.chromatogr. (A), vol.1034, pp. 161-167, 2004. |
| [4] | L.Zhou, Bruce D.Johnson, C.Miller and Jean M.Wyvratt, “The separation of positional isomers by capillary electrochromatography”,J. chromatogr. (A), vol. 875, pp. 389-401, 2000. |
| [5] | D.D.Schumacher, C.R. Mitchell, T. Xiao, R.V.Rozhkov, R.C. Larock and D. W. Armstrong, “Determination of polyethylene glycol in low-density polyethylene by large volume injection temperature gradient packed capillary liquid chromatography”, J. chromatogr. (A), Vol.1011, pp.37-47, 2003. |
| [6] | J-Y Moon, H-J Jung, M. H. Moon, B.C. Chung and M.H. Choi, “Inclusion complex-based solid-phase extraction of steroidal compounds with entrapped β-cyclodextrin polymer”, Steroids (A), Vol.73, pp.1090-1097, 2008. |
| [7] | C-H Lin,W-R Fang ,C-M Kuo,W-Y Chang,Y-C Liu ,W-Y Lin ,J-C Wu and C-E Lin , “Chiral separation of hydroxyflavanones in cyclodextrin-modified capillary zone electrophoresis using sulfated cyclodextrins as chiral selectors”,J. chromatogr. (A), Vol.1188, pp.301-307, 2008. |
| [8] | Y.Francois, A.Varenne, E. Juillerat, D.Villemin and P. Gareil, “Evaluation of chiral ionic liquids as additives to cyclodextrins for enantiomeric separations by capillary electrophoresis”, J. chromatogr. (A), Vol.1155, pp.134-141, 2007. |
| [9] | V.Cucinotta, A.Giuffrida, G.Grasso, G.Maccarrone, M.Messina and G.Vecchio, “High selectivity in new chiral separations of dansyl amino acids by cyclodextrin derivatives in electrokinetic chromatography”, J.chromatogr. (A), Vol.1155, pp.172-179, 2007. |
| [10] | H-D Wang, L-Y Chu, H.Song, J.P.Yang, R. Xie and M. Yang, “Preparation and enantiomer separation characteristics of chitosan/ β-cyclodextrin composite membranes”, J.Membrane Sc., Vol.297,pp-262-270, 2007. |
| [11] | C. Jullian, S. Miranda, G. Zapata-Tarres, F. Mendizabal, C. Olea-Azar, “Studies of inclusion complexes of natural and modified cyclodextrin with (+) catechin by NMR and molecular modeling”, Bioorganic & Medicinal Chemistry, Vol.15, pp.3217-3224, 2007. |
| [12] | P.Neoff, U, F.Haustein and N.Hittel, “Activity of Nadifloxacin (OPC-7251) and Seven Other Antimicrobial Agents against Aerobic and Anaerobic Gram-Positive Bacteria Isolated from Bacterial Skin Infections”, Int. J. of Experimental and Clinical Chemotherapy, Vol.50,pp.196-198, 2004. |
| [13] | United States food and drug administration, Chirality,Vol.4, pp-338-340, 1992. |
| [14] | D.W.Armstrong and W.Demond, “Enantiomeric resolution of dansyl phenylalanine and analogs by microcolumn liquid chromatography withβ-cyclodextrin as mobile phase additive”, J .Chromatogr .Sc., Vol.22, pp.411-415, 1984. |
| [15] | D.W.Armstrong, T.J.Word, R.D.ArmstrongandT.F.Beesley, “Separation of drug stereoisomers by the formation of beta-cyclodextrin inclusion complexes”, Science, Vol.232, pp.1132-1142. 1986. |
| [16] | C.R.Mitchell, M.Desai, R.Mcculla, W.Jenks andD.W.Armstrong, “Use of Native and DerivatizedCyclodextrin Chiral Stationary Phases for the Enantioseparation of Aromatic and Aliphatic Sulfoxides by High Performance Liquid Chromatography”, Chromatographia, Vol.56, pp.127-132, 2002. |
| [17] | D.W.Armstrong and J.Jukoski, “Direct enantiomeric resolution of monoterpene hydrocarbons via reversed-phase high-performance liquid chromatography with an α-cyclodextrin bonded stationary phase”, J.Chromatogr. (A), Vol.666, pp.445-448, 1994. |
| [18] | D.W.Armstrong, T.L.Ward, A.Czech, B.P.Czech and R.A.Bastsch., “Synthesis, rapid resolution, and determination of absolute configuration of racemic 2,2'-binaphthyldiyl crown ethers and analogsvia .beta.-cyclodextrincomplexation”, J.Org.Chem., Vol.50, pp.5556-5559, 1985. |
| [19] | D.W.Armstrong, L.W.Chang, S.C.Chang, X.Wang, H.Ibrahim, G.R.Reid and T.E.Beesley, “Comparison of the Enantioselectivity of β-Cyclodextrin vs.Heptakis-2,3-O-dimethyl-β-cyclodextrin LC Stationary Phases Comparison of the Enantioselectivity of β-Cyclodextrin vs.Heptakis-2,3-O-dimethyl-β-cyclodextrin LC Stationary Phases”, J.Liq. Chromatogr.Rel.Technol, Vol.20, pp.3279-3295, 1997. |
| [20] | D.W.Armstrong, W.Demond and B.P.Czech., “Separation of Metallocene Enantiomers by Liquid Chromatography Chiral Recognition Via Cyclodextrin Bonded Phases”, Anal chem., Vol.57,pp.481-484, 1985. |
| [21] | M.L. Bender and M.Komiyama, cyclodextrin chemistry, Springer, Berlin. 1978. |
| [22] | J.J. Tang, Dissertation, Seton Hall University, N.J., 1996. |
| [23] | J.J. Tang and L.J.Clinelove, “Formation constants of polynuclear aromatic compounds and β-cyclodextrin inclusion complexes in β-cyclodextrin modified mobile phase high performance liquid chromatography system”, Anal.chim.Acta, Vol.344, pp.137-143, 1997. |
| [24] | J.I.Seeman, H.V.Secor, D.W. Armstrong, K.D.Timmons and T.J. Ward, “Enantiomeric resolution and chiral recognition of racemic nicotine and nicotine analogsby .beta.-cyclodextrincomplexation. Structure-enantiomeric resolution relationships in host-guest interactions”, Anal.chem., Vol.60, pp-2120-2127, 1988. |
| [25] | W.L. Hinze, T.E Riehl, D.W.Armstrong, W.Demond, A.Alak and T.J.Ward, “Liquid chromatographic separation of enantiomers using a chiral beta.-cyclodextrin-bonded stationary phase and conventional aqueous-organic mobile phases”, Anal.Chem., Vol.57,pp-237-242, 1986. |
| [26] | H.Yang, Ph.D Thesis, University of Pitts burgh., pp.33-34, 2008. |