-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Advances in Analytical Chemistry
p-ISSN: 2163-2839 e-ISSN: 2163-2847
2012; 2(3): 6-13
doi: 10.5923/j.aac.20120203.01
Comparison of High Performance Liquid Chromatography and Three Titrimetric Methods for the Determination of Ceftazidime in Pharmaceutical Formulations
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLAndréia de Haro Moreno, Hérida Regina Nunes Salgado
Department of Drugs and Medicines, School of Pharmaceutical Sciences, University of São Paulo State, Araraquara, 14801-902, Brazil
Correspondence to: Hérida Regina Nunes Salgado, Department of Drugs and Medicines, School of Pharmaceutical Sciences, University of São Paulo State, Araraquara, 14801-902, Brazil.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Three different assays for the quality control of ceftazidime in commercial formulations were developed and compared: acidimetric, iodometric and nonaqueous methods. Method validation yielding good results and included precision and accuracy, with good recovery percent ranged from 99.67 to 100.39, and R.S.D. values smaller than 2%. Results were compared to those obtained by high performance liquid chromatographic method, developed and published previously by us, and no evidence of significant difference was observed. The results obtained agreed well with the contents stated on the labels, being rapid, simple and inexpensive alternative method for the determination of ceftazidime in pharmaceutical formulations.
Keywords: Ceftazidime, HPLC, Method Comparison, Pharmaceutical Analysis, Quality Control, Titrimetry
Article Outline
1. Introduction
- Ceftazidime is a semisynthetic cephalosporin of the third generation with high antibacterial activity, widely used in the treatment of commonly-occurring bacterial infections, including indole-positive Proteus species and Pseudomonas aeruginosa and have been considered to be the drugs of choice for serious infections caused by Klebsiella, Enterobacter, Proteus, Providencia, Serratia and Haemophylus species[1-7]. They include biliary-tract infections, bone and joint infections, cystic fibrosis (respiratory-tract infections), endophthalmitis, infections in immunocompromised patients (neutropenic patients), meningitis, peritonitis, pneumonia, septicaemia, skin infections (including burns, ulceration) and urinary-tract infections[1,8-11]. Favorable properties of ceftazidime include efficient penetration of the bacterial cell wall, resistance to bacterial enzyme degradation, a high intrinsic activity against the bacterial cell targets, a broad spectrum of activity, very low toxicity, extensive tissue penetration, metabolic stability, and a low degree of serum protein binding[9,12-14]. The chemical structure of ceftazidime is represented by Figure 1. Ceftazidime is administrated by injection as the sodium salt or in solution with arginine and is widely distributed in body tissues and fluids; it crosses the placenta and is distributed into breast milk[15]. Penetration into the aqueous humor of the eye is relatively good after systemic administration of ceftazidime[1,16,17]. There is some evidence that concentrations sufficient for therapy of ocular infections due to gram-positive and certain gram-negative microorganisms can be achieved after systemic administration[15].
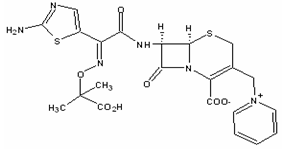 | Figure 1. Chemical structure of ceftazidime – C22H22N6O7S2 (mw 546.58) |
2. Material and Methods
2.1. Chemicals
- Ceftazidime reference substance (assigned purity 99.98%) and ceftazidime powder for injection were kindly supplied by Ariston Química e Farmacêutica Ltda. (). Ceftazidime powder for injection (Ceftazidon) was claimed to contain 1000 mg (as anhydrous base) of the drug and 118 mg of anhydrous sodium carbonate as excipient (solubilizer).Solutions were made according to USP Pharmacopeia 2008 (45). All chemicals used were from analytical grade.a) hydrochloric acid: 8.5 mL of hydrochloric acid (Merck, Darmstadt, Germany) were diluted with water to 1000 mL. This solution was standardized as following: of sodium carbonate (Merck, Darmstadt, Germany) previously crushed lightly and dried at 270℃ for 1 h) were accurately weighed and dissolved in 100 mL of water. So, 2 drops of methyl red solution were added and titrated with the hydrochloric solution to the production of a permanent pale pink color. Each 52.99 mg of sodium carbonate is equivalent to 1 mL of hydrochloric acid.b) hydrochloric acid: 85.0 mL of hydrochloric acid (Merck, Darmstadt, Germany) was diluted with water to 1000 mL; c) iodine: of iodine was dissolved in an aqueous potassium iodide solution (36%, w/v) and 3 drops of hydrochloric acid was added; so, this solution was diluted with water to 1000 mL, and standardized as United States Pharmacopeia (45). An aliquot of 50 mL of this solution was diluted in water to make 1000 mL; d) 0.1 N perchloric acid: 8.5 mL of perchloric acid (Merck, Darmstadt, Germany) were mixed with 500 mL of glacial acetic acid (Merck, Darmstadt, Germany) and 21 mL of acetic anhydride (Merck, Darmstadt, Germany), cooled, and added glacial acetic acid to make 1000 mL. This solution was standardized as follows: 700 mg of potassium biphthalate were accurately weighed (previously dried at 120℃ for 2 h) and dissolved in 50 mL of glacial acetic acid in a flask. So, 2 drops of crystal violet solution were added, and titrated with perchloric acid solution until blue-green color. Each 20.42 mg of potassium biphthalate is equivalent to 1 mL of 0.1 N perchloric acid.e) sodium hydroxide: of sodium hydroxide (Merck, Darmstadt, Germany) was dissolved in 150 mL of carbon dioxide-free water, cooled at room temperature and diluted with carbon dioxide-free water to 1000 mL. This solution was standardized as following: of potassium biphthalate (previously dried at 120℃ for 2 h) were accurately weighed and dissolved in 75 mL of carbon dioxide-free water in a flask. After, 2 drops of phenolphthalein solution were added, and titrated with the sodium hydroxide solution to the production of a permanent pink color. Each 20.42 mg of potassium biphthalate is equivalent to 1 mL of sodium hydroxide.f) sodium hydroxide: of sodium hydroxide (Merck, Darmstadt, Germany) was dissolved in water to make 1000 mL;g) sodium thiosulphate: of sodium thiosulphate (Merck, Darmstadt, Germany) was dissolved in water to make 1000 mL. An aliquot of 50 mL of this solution was diluted in water to make 1000 mL; h) Acetate buffer solution: of sodium acetate (Merck, Darmstadt, Germany) and of glacial acetic acid were dissolved in water to make 100 mL.i) Crystal violet 1% (w/w): 100 mg of crystal violet (Merck, ) were dissolved to 10 mL of glacial acetic acid.j) Methyl red 0.1% (w/w): 100 mg of methyl red (Merck, Darmstadt, Germany) were dissolved in 100 mL of alcohol and filtered.k) Phenolphthalein 1% (w/w): 100 mg of phenolphthalein (Merck, Darmstadt, Germany) were dissolved in 100 mL of alcohol and filtered.l) Starch mucilage: of soluble starch was mixed with 10 mg of red mercuric iodide and sufficient cold water to make a thin paste. So, 200 mL of boiling water was added with continuous stirring. After cool, the clear solution was used.
2.2. Procedure
- In order to determine the average weight, contents of 20 flasks of ceftazidime powder for injection (vials) were individually weighed and mixed.a) Acidimetric method: an accurately weighed portion of the power equivalent to 500 mg of the drug was transferred in a 250 mL conical flask and dissolved with 25 mL of carbon dioxide-free water (previously neutralized with hydrochloric acid). An aliquot of 25.0 mL of sodium hydroxide was added and heated in a water bath at 80℃ for 20 minutes. This time was found appropriate for complete hydrolysis of cephalosporins[22]. The solution was cooled at room temperature, and 2 drops of phenolphthalein solution were added and the excess of sodium carbonate was titrated with hydrochloric acid. A blank determination was performed and the difference between titrations represented the volume of sodium hydroxide equivalent to quantity of ceftazidime present. This procedure was performed in triplicate. Each mL of sodium hydroxide is equivalent to 54.658 mg of ceftazidime (as anhydrous base).b) Non aqueous method: An accurately weighed portion of powder equivalent to 250 mg of drug was dissolved in water and transferred to a 250 mL conical flask, dissolved with 30 mL of glacial acetic acid and 2 drops of crystal violet solution were added and mixed. Drug was titrated with 0.1 N perchloric acid until the blue-green color. A blank determination was performed at 20℃ and any necessary corrections were made. This procedure was performed in triplicate. Each mL of 0.1 N perchloric acid is equivalent to 54.658 mg of ceftazidime (as anhydrous base).c) Iodometric method: An accurately weighted portion of the power equivalent to 100 mg of the drug was dissolved in water and transferred in a 100 mL volumetric flask (1000 µg/mL). An aliquot of 10 mL was transferred to a stopped flask, added 5 mL of sodium hydroxide, and allowed to stand for twenty minutes. So, 20 mL of a freshly prepared acetate buffer solution, 5 mL of hydrochloric acid, and 25 mL of iodine were added and the flask closed with a wet stopper. After 20 minutes, protected from light, the excess of iodine was titrated with sodium thiosulphate, using starch mucilage, added towards the end of the titration, as indicator. To a further 10 mL of the ceftazidime solution (1000 µg/mL) was added 20 mL of the acetate buffer solution and 25 mL of iodine, allowed to stand for twenty minutes and titrated with sodium thiosulphate, using starch mucilage, added towards the end of the titration, as indicator. A blank determination was performed at a temperature of 20℃ and any necessary corrections were made. The difference between titrations represents the volume of iodine equivalent to the ceftazidime present. Each mL of iodine is equivalent to 1.5915 mg of ceftazidime (as anhydrous base).
2.3. Method validation
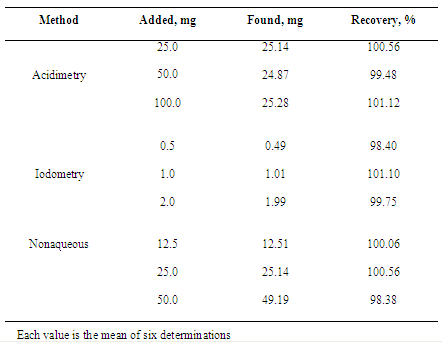
- The three titrimetric methods were validated by the following parameters: precision and accuracy. The recovery percentage of ceftazidime added was calculated using the equation proposed by AOAC[52].a) Precision: Repeatability (intraday) and intermediate precision (interday) of the proposed assay were evaluated. Repeatability was studied by the assay of independent samples, during the same day under the same experimental conditions. Intermediate precision was evaluated by comparing the results obtained on 3 different days. Precision was expressed as percent coefficient of variation. Analysis of variance (ANOVA) is an important statistical tool to verify the internal validity of an analytical procedure.b) Accuracy: This parameter was determined by the recovery study, comparing the theoretical and measured concentrations of known amounts of ceftazidime reference substance added at the beginning of the process. For the acidimetric method, the equivalent to 5000 mg of ceftazidime reference substance was dissolved in sufficient water to produce 500 mL (10.0 mg/mL). Amounts of ceftazidime powder for injection (equivalent to 500 mg of the drug) were transferred to 250 mL conical flasks (R1, R2 and R3) and dissolved with 25 mL of carbon dioxide-free water (neutralized with hydrochloric acid). Portions of 2.5, 5.0 and 10.0 mL of ceftazidime reference substance (10.0 mg/mL) were added to flasks R1, R2 and R3, respectively. Aliquots of 25.0 mL of sodium hydroxide were added and heated in a water bath at 80℃ for 20 minutes. After the solutions were cooled at room temperature, 2 drops of phenolphthalein solution were added in each flask and the excess of sodium hydroxide was titrated with hydrochloric acid. A blank determination was performed and the difference between the titrations represented the volume of sodium hydroxide equivalent to ceftazidime present. This procedure was performed in triplicate. Each mL of sodium hydroxide is equivalent to 54.658 mg of ceftazidime (as anhydrous base).For nonaqueous method, the equivalent to 2500 mg of ceftazidime reference substance was dissolved in sufficient glacial acetic acid to produce 500 mL (5.0 mg/mL). Amounts of ceftazidime powder for injection (equivalent to 250 mg of the drug) were transferred to 250 mL conical flasks (R1, R2 and R3) and dissolved with 30 mL of glacial acetic acid. Portions of 2.5, 5.0 and 10.0 mL of ceftazidime reference substance (5.0 mg/mL) were added to flasks R1, R2 and R3, respectively. After, 2 drops of crystal violet solution were added in each flask and the solutions were titrated with 0.1 N perchloric acid until the blue-green color. A blank determination was performed at 20℃ and any necessary corrections were made. This procedure was performed in triplicate. Each mL of 0.1 N perchloric acid is equivalent to 54.658 mg of ceftazidime (as anhydrous base).For iodometric method, recoveries were determined by adding known amounts of ceftazidime reference substance (0.5, 1.0 and 2.0 mg) to the samples at the beginning of the procedure. Ceftazidime reference substance and powder for injection were dried in vacuum at a pressure not exceeding of mercury at 60℃ for 3 hours. The equivalent of 125 mg ceftazidime reference substance was dissolved in sufficient water to produce 25 mL (5 mg/mL). Amounts of 1.0, 2.0 and 4.0 mL of this solution were added with the sample (aliquot of 10 mL) and transferred to a stopped flask, added 5 mL of sodium hydroxide, and allowed to stand for 20 minutes. After, 20 mL of a freshly prepared acetate buffer solution, 5 mL of hydrochloric acid, and 25 mL of iodine were added and the flask closed with a wet stopper, and allowed to stand for 20 minutes, protected from light. Excess of iodine was titrated with sodium thiosulphate, using starch mucilage, added towards the end of the titration, as indicator. To a further 10 mL of ceftazidime solution (1000 µg/mL) was added 20 mL of acetate buffer solution and 25 mL of iodine, allowed to stand for twenty minutes and titrated with sodium thiosulphate, using starch mucilage, added towards the end of the titration, as indicator. The difference between titrations represents the volume of iodine equivalent to the ceftazidime present.c) Method comparison: Results obtained in this study were compared to those obtained by a high performance liquid chromatography method, previously developed and validated by us[49].
3. Results and Discussion
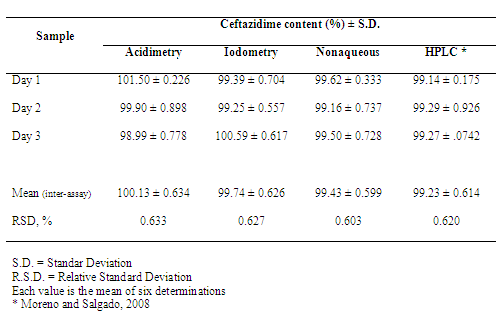
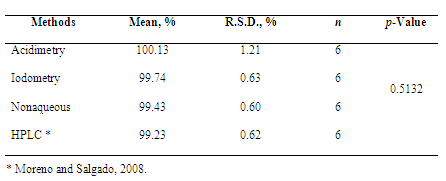
- Acidimetric methodCephalosporins are antibiotics widely used to the treatment of various bacterial infections. The minimum requirement for the activity of this drug group is the presence of the beta-lactam ring connected to the acid group (Figure 1). Most chemical methods of assay for the cephalosporins depend upon hydrolytic cleavage of the β-lactam ring to give cephalosporanic acid.Antibiotic hydrolytic degradation is frequently used as preliminary stage in the quantification analytical procedures, being the alkaline hydrolysis the most common[24,53]. According to Korolkovas[53], penicillins and cephalosporins can be determined by alkaline hydrolysis of beta-lactam ring using known accurately amount of sodium hydroxide solution, providing the sodium salt of penicilloic acid derived. After, excess of sodium hydroxide solution is quantified by standardized acid solution (generally hydrochloric acid).Ceftazidime commercial sample used in this study (powder for injection) presented in its composition sodium carbonate as excipient (solubilizer), equivalent to 2.3 mEq Na+ in 10 mL of reconstituted sample. This excipient interferes with the assay (also reacts with the acid solution used to quantify sodium hydroxide excess). So, this interference was eliminated by sample neutralization with hydrochloric acid solution at room temperature before alkaline hydrolysis (that occurs generally by heat). This procedure not provided interference in the calculations, according to the results obtained and values subsequently presented by the recovery study.Experimental values obtained for ceftazidime powder for injection are presented in Table 1, showing the agreement between the accepted value and the value found to be 100.13% for powder for injection.Precision and accuracy of the assay were also demonstrated. Precision is usually expressed as the variance, relative standard deviation (R.S.D.), or coefficient of variation (C.V.%) of a series of measurements[54]. Results obtained through the acidimetric assay in 3 different days showed a R.S.D. less than 2% (0.633%), indicating good repeatability of the assay (Table 1).Accuracy was measured by the recovery test, which was obtained by comparing the experimental values to the calculated theoretical concentrations. The mean absolute recovery test of acidimetric assay was found to be 100.39 ± 0.83%, indicating a good accuracy and the agreement with the spiked amount of reference substance (Table 2).Pharmacopeias don’t recommend acidimetric assay for cephalosporins; so, the proposed assay showed simple, accessible and unpublished for the quantification of ceftazidime. Data obtained by the proposed assay were statistically compared by ANOVA test with the one obtained by HPLC method previously developed and validated by us (Table 3). It was indicated that there is no evidence of significant difference between these methods (p<0.05). For this reason, the proposed assay showed appropriate for the determination of ceftazidime in powder for injection and can be used in routine quality control analysis.
|
|
|
4. Conclusions
- We could conclude that the acidimetric, nonaqueous and iodometric assays proposed are simple, rapid and inexpensive and can therefore be applied to the determination of ceftazidime in raw material and pharmaceuticals. Method validation yielding showed good results and included precision and accuracy. It was found that the excipient interference was eliminated, according to the results obtained. Results of statistic analysis showed this assay gives similar results to those obtained by HPLC, indicating that it allows reliable determination of ceftazidime and can be used for the quality control analysis.
ACKNOWLEDGEMENTS
- Authors thank Ariston Química e Farmacêutica Ltda. (São Paulo, Brasil) for providing ceftazidime reference substance and ceftazidime powder for injection. This work was supported by PACD-FCFAr-UNESP-Brazil, FUNDUNESPBrazil, FAPESP-Brazil and CNPq-Brazil.